

一种简便易行的利用全基因组数据构建进化树的方法
随着测序变得越来越普遍越来越便宜,很多科研人员都开始尝试测一测自己关心的物种的基因组。对于生态学家来讲,关心的物种千奇百怪,且多是前人没有测过的,相当于没有一个模版。测出来的数据长则几百,短则几十,零碎的不行,在没有模版的情况下很难组装成一个完整的基因组,便也难起大的作用。
针对现阶段测序技术测出来的片段较短,已组装成型的基因组较少的现状,版纳植物园动植物关系组的博士研究生范欢在导师Tony Ives, YannSurget-Groba和Chuck Cannon的指导下,开发了名为Alignment and assembly-free (AAF)的软件包,用于直接分析测序仪上下来的原始数据,不经组装和比对,通过原始序列里短序列的相似度计算基因组之间的遗传距离,然后用以构建进化树。之前也有很多不经比对或者同时不经组装就构建进化树的方法,AAF最大的优势在于它并行处理大规模数据的能力。处理人类基因组大小的基因组产生的测序数据亦不在话下。同时它还拥有修复测序错误和计算bootstrap值的功能,是一款实实在在为用户考虑的软件。
该研究已以An assembly and alignment-free method of phylogeny reconstruction from next-generation sequencing data为题发表在BMC Genomics上,发表后被标为“Highly accessed”文章。该软件可于以下网址免费下载。软件包中含有使用手册和测试数据。该软件除用于全基因组测序数据外,也可用于其他类型的下一代测序数据例如RAD-seq. 针对RAD-seq的软件包尚未发表,如有需要请联系fh@xtbg.org.cn
AAF下载地址:https://sourceforge.net/projects/aaf-phylogeny
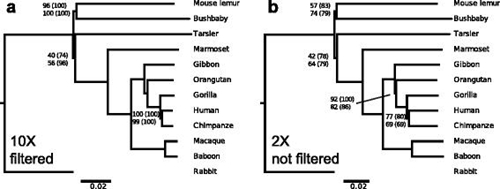
用AAF处理灵长类基因组数据后得到的进化树,未标注的节点表明100%的支持率。



滇ICP备13004273号 滇公网安备53282302000011号


今日头条

官方微博

微信公众号
官方抖音号